

The main focus of our research laboratory is to determine the causes of thyroid autoimmune disease with the TSH receptor as the key molecule of interest. The TSH receptor (TSHR) is a G protein-coupled receptor on the basolateral surface of the thyrocytes and which regulates thyroid growth and metabolism. The TSHR is also a major auto antigen in human autoimmune thyroid disease such as Graves’ disease (autoimmune hyperthyroidism).
Our laboratory studies various aspects of the TSHR including its genetic association with disease, the multiple post-translational changes it undergoes and the important role, of TSHR antibodies in thyroid autoimmune disease. The laboratory is also developing novel small molecule therapeutics against the TSH receptor. In addition, we have developed major activity in the area of human thyroid stem cells and the role of extra thyroidal TSHRs found in bone osteoclasts and osteoblasts.
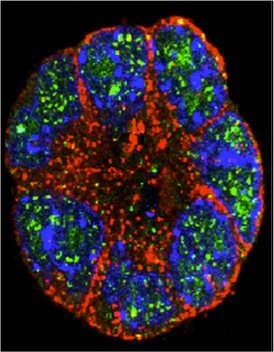
Thyroid follicle from embryonic stem cells.

TSH receptor “knock out” mouse.